Прогрессирующая мышечная дистрофия Дюшенна

Прогрессирующая мышечная дистрофия Дюшенна — наследуемая сцеплено с Х-хромосомой патология мышечной системы, проявляющаяся в первые 3-5 лет жизни и характеризующаяся быстро распространяющейся и усугубляющейся мышечной слабостью. Первоначально поражаются мышцы тазового пояса и бедер, затем — плеч и спины, постепенно наступает обездвиженность. Миодистрофия сопровождается скелетными деформациями и поражением сердца. Диагностика дистрофии Дюшенна включает неврологическое и кардиологическое обследование, определение уровня КФК, электромиографию, консультацию генетика, ДНК-анализ, биопсию мышц. Лечение симптоматическое. В связи со слабостью дыхательной мускулатуры на заключительном этапе заболевания требуется ИВЛ.
- Причины
- Симптомы
- Осложнения
- Диагностика
- Лечение мышечной дистрофии Дюшенна
- Стандартная терапия
- Экспериментальная терапия
Общие сведения
Прогрессирующая мышечная дистрофия Дюшенна — тяжелая форма миодистрофии, отличающаяся ранним началом, быстрым усугублением мышечной слабости, выраженными деформациями скелета и поражением сердечной мышцы. Впервые была описана французским неврологом Дюшенном в 1853 году. Ее распространенность составляет 1 случай на 4 тыс. новорожденных мальчиков. Патология передается рецессивно сцеплено с Х-хромосомой. Заболевают мальчики. Известны случаи заболевания среди девочек, что связано с кариотипом ХО, гонадотропным мозаицизмом или наличием аномалий в структуре хромосом. Миодистрофия Дюшенна характеризуется началом в первые 3-5 лет жизни ребенка, тяжелым течением, приводящим к полной обездвиженности и гибели пациентов в среднем к возрасту 15-25 лет.
Прогрессирующая мышечная дистрофия Дюшенна
Причины
Развитие мышечной дистрофии Дюшенна связано с наличием мутации в 21-ом локусе короткого плеча Х-хромосомы в гене, кодирующем белок дистрофин. Около 70% случаев болезни вызваны дефектным геном дистрофина, полученным от матери — носительницы патологической мутации. Остальные 30% связаны с появлением свежих мутаций в яйцеклетках матери. В отличие от миодистрофии Беккера, при дистрофии Дюшенна генетические аберрации приводят к сдвигу рамки считывания ДНК и полному прекращению синтеза дистрофина, что и обуславливает более тяжелое течение патологии.
В норме входящий в сарколемму миоцитов дистрофин обеспечивает ее целостность и устойчивость к растяжению, возникающему при сократительной активности мышечных волокон. Отсутствие дистрофина влечет за собой нарушение целостности сарколеммы, разрушение миоцитов и их замещение жировой и соединительной тканью. Клинически этот процесс выражается прогрессирующим снижением способности мышц к сокращению, утратой мышечной силы и тонуса, атрофией мышц.
Симптомы
Дебют миодистрофии Дюшенна приходится на период от 1 до 5 лет. Как правило, уже на 1-ом году жизни заметно некоторое отставание моторного развития ребенка. Отмечается задержка сроков начала сидения, самостоятельного вставания и ходьбы. Когда ребенок начинает ходить, он отличается неуклюжестью и большей, по сравнению со сверстниками, неустойчивостью; часто спотыкается.
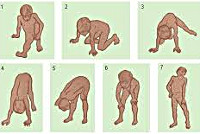
Мышечная слабость возникает на 3-4-ом годах жизни. Первоначально она выражается в патологически повышенной утомляемости при ходьбе по лестнице или на длинные расстояния. Со временем становится заметной типичная для миодистрофий утиная походка. Обращают на себя внимание особенности поведения ребенка — каждый раз, поднимаясь из положения сидя на корточках, он активно опирается руками о собственное тело, как бы взбираясь по нему как по лесенке (симптом Говерса).
Мышечные атрофии начинаются с мышц бедер и тазового пояса. Для дистрофии Дюшенна характерно их быстрое восходящее распространение на плечевой пояс, мускулатуру спины и проксимальных отделов рук. Вследствие мышечных атрофий формируется «осиная» талия и отстоящие от спины «крыловидные» лопатки. Типичным симптомом выступает псевдогипертрофия икроножных мышц. Наблюдается выпадение сухожильных рефлексов: вначале — коленных, затем — рефлексов с трицепса и бицепса плеча. Ахилловы и карпорадиальные рефлексы могут длительное время быть сохранны. Со временем развиваются ретракции сухожилий и мышечные контрактуры.
Прогрессирующая мышечная дистрофия Дюшенна сопровождается нарушениями в костно-суставной системе. Характерны искривление позвоночника (кифоз, усиленный лордоз, сколиоз), деформации грудной клетки (килевидная или седловидная), деформации стоп. Сердечно-сосудистые расстройства обусловлены развитием кардиомиопатии и включают аритмию, лабильность артериального давления, глухость тонов сердца. У 50% больных фиксируются нейроэндокринные расстройства — адипозогенитальная дистрофия, синдром Иценко-Кушинга и др. Около 30% больных страдает олигофренией, как правило, ограничивающейся степенью дебильности. Могут отмечаться СДВГ, расстройства по типу аутизма, дислексия, нарушения краткосрочной памяти.
Осложнения
Уже к 7-10-летнему возрасту дистрофия Дюшенна приводит к выраженным двигательным ограничениям. К 12 годам больные, как правило, утрачивают способность ходить, а к возрасту 15 лет большинство пациентов полностью теряют возможность самостоятельных движений. Распространение дистрофического процесса на дыхательную мускулатуру приводит к прогрессирующему падению жизненной емкости легких (ЖЕЛ) и, в конечном итоге, невозможности совершать дыхательные движения.
Диагностика
Установить диагноз миодистрофии Дюшенна помогает анамнез, неврологическое обследование, результаты электрофизиологического тестирования, определение креатинфосфокиназы (КФК) в биохимическом анализе крови, морфологическое и иммунохимическое исследование образцов мышечной ткани, генетическое консультирование и анализ ДНК:
- ЭФИ. Электронейро- и электромиография определяют сохранность проведения импульсов по нервным волокнам, пониженную амплитуду М-ответа, что свидетельствует о первично-мышечном типе поражения. Характерным является 30-50-кратный подъем уровня креатинфосфокиназы.
- Генетическая диагностика. На консультации генетика проводится генеалогическое исследование, позволяющее выявить наличие случаев миодистрофии Дюшенна в семье больного и определить женщин, являющихся носительницами мутантного гена дистрофина. Диагностика ДНК позволяет выявить аномалии в гене дистрофина. Следует учитывать, что невыявление мутации при ДНК-анализе не говорит о ее отсутствии, поскольку поиск точковых мутаций обычно не входит в задачи анализа из-за его большой длительности и трудоемкости.
- Биопсия. В случаях, когда имеется клиническая картина миодистрофии, а анализ ДНК не выявил наличие мутации, показана биопсия мышц. Морфологическое исследование биоптата определяет разнокалиберность и некроз миоцитов, их замещение соединительнотканными элементами. Иммунохимический анализ говорит о полном отсутствии дистрофина в исследуемых мышечных волокнах.
- Другие исследования. Дополнительно осуществляется обследование костно-мышечной и сердечно-сосудистой систем — проводится консультация ортопеда, рентгенография позвоночника, обзорная рентгенография ОГК, консультация кардиолога, ЭКГ, эхокардиография. По показаниям рекомендуется консультация эндокринолога, пульмонолога и др. специалистов.
При этом дифференциальную диагностику следует проводить с другими миопатиями — метаболической, воспалительной, миодистрофией Беккера, мышечной дистрофией Дрейфуса, дистрофией Эрба-Рота, а также с полиневропатиями, полимиозитом, БАС.
Лечение мышечной дистрофии Дюшенна
Стандартная терапия
Терапия, применяемая в клинической практике, включает симптоматическое и патогенетическое направление. В рамках данных направлений применяется медикаментозная терапия, физическая реабилитация, респираторная поддержка:
- Кортикостероиды. Основная роль в лечении мышечной дистрофии Дюшенна на сегодняшний день отводится глюкокортикостероидам, которые назначаются как способным, так и не способным к самостоятельному передвижению пациентам. ГКС помогают замедлить прогрессирование мышечной слабости, оказывают умеренный пульмопротективный и кардиопротективный эффект, снижают риск развития ортопедических осложнений. Из-а большого количества побочных эффектов глюкокортикостероидной терапии необходим тщательный мониторинг состояния ребенка, своевременная коррекция дозы и схемы приема препарата.
- Метаболическая терапия. Направлена на улучшение обменных процессов в скелетной мускулатуре, костях, сердечной мышце, печени, снижение побочных эффектов от приема ГКС. Включает назначение витаминов группы В, левокарнитина, препаратов Са, витамина D.
- Физическая терапия. С целью минимизировать образование контрактур и продлить двигательную активность пациентов проводится ЛФК, массаж, физиотерапия, пассивная и активная растяжка. Рекомендуется использование ортезов, вертикализатора, специальных шин, занятия лечебным плаванием.
- Респираторная поддержка. Важное значение имеет контроль дыхательной функции и газового состава крови. При падении ЖЕЛ до 40% рекомендована искусственная вентиляция легких в период сна. В дальнейшем время ИВЛ растет пропорционально снижению ЖЕЛ. В начале ИВЛ может осуществляться при помощи масочного аппарата. Затем необходима трахеостомия, и ИВЛ проводится путем присоединения аппарата к трахеостомической трубке. Современные портативные аппараты ИВЛ работают на батареях и могут быть закреплены на инвалидной коляске.
Экспериментальная терапия
Перспективным направлением является разработка и использование препаратов, нацеленных на восстановление синтеза или замещение дистрофина. В 2020 г. в России был зарегистрирован первый таргетный препарат для лечения мышечной дистрофии Дюшенна — аталурен, который подходит 10% пациентам с нонсенс-мутацией (стоп-кодоном). Препарат позволяет возобновить синтез белка дистрофина в организме, требует пожизненного приема. Другие аналогичные лекарственные средства (этеплирсен, голодирсен) в РФ пока не одобрены к применению. Положительные результаты на стадии клинических испытаний у пациентов с миодистрофией Дюшенна показал микродистрофин. Идет поиск и апробация методов заместительной генной терапии.
Прогноз и профилактика
Из всех форм миодистрофии дистрофия Дюшенна имеет наиболее неблагоприятный прогноз. Манифестация заболевания в раннем возрасте приводит к тому, что к 15 годам пациенты становятся полностью обездвижены. Летальный исход неизбежен. Зачастую больные не достигают 25-летнего возраста. Обычно смертельный исход обусловлен интеркуррентными инфекциями, застойной пневмонией, сердечной или дыхательной недостаточностью.
Профилактические мероприятия направлены на выявление женщин-носительниц аномального гена дистрофина и предупреждение рождения у них больного ребенка. В рамках профилактических мер проводятся консультации генетика для планирующих беременность супружеских пар, консультации беременных и пренатальная ДНК-диагностика.
Источник https://www.krasotaimedicina.ru/diseases/zabolevanija_neurology/Duchenne-muscular-dystrophy